

github:地址
Rdkit|分子性质描述符(Descriptors)
性质描述符计算
rdkit.Descriptors 模块可以计算许多分子属性,包括但不限于以下几个方面:
物理化学性质:
- 分子量(Molecular weight)
- 分子式(Molecular formula)
- 荷电数(Formal charge)
- 荷电状态(Formal charge state)
- 碳原子数(Number of carbon atoms)
- 氢键供体数(Number of hydrogen bond donors)
- 氢键受体数(Number of hydrogen bond acceptors)
- 旋光度(Optical rotation)
- 紫外吸收最大波长(Maximum UV absorbance wavelength)
拓扑性质:
- 莫尔体积(Molar volume)
- 莫尔极性表面积(Molar polar surface area)
- 倍数不饱和度(Ring count)
- 原子数(Number of atoms)
- 原子种类数(Number of atom types)
- 键数(Number of bonds)
- 键种类数(Number of bond types)
几何性质:
- 分子长度、宽度和高度(Molecular length, width, and height)
- 分子形状描述符(Molecular shape descriptors)
- 分子表面积(Molecular surface area)
药物性质:
- 脂溶性(LogP)
- 水溶性(LogS)
- 生物利用度(Bioavailability)
- 药物相似性(Drug likeness)
电子结构性质:
- 能量最低的分子轨道(Lowest energy molecular orbital)
- 能量最高的分子轨道(Highest energy molecular orbital)
- 能量最低的半满分子轨道(Lowest energy half-filled molecular orbital)
- 能量最高的非满分子轨道(Highest energy non-filled molecular orbital)
分子动力学性质:
- 热力学性质(Thermodynamic properties)
- 动力学性质(Kinetic properties)
- 活性位点描述符(Active site descriptors)
其他属性:
- 拓扑极性表面积(Topological polar surface area)
- 晶体密度(Crystal density)
- 分子内氢键数量(Number of intramolecular hydrogen bonds)
- 分子间氢键数量(Number of intermolecular hydrogen bonds)
- 分子内离子对数量(Number of intramolecular ion pairs)
- 分子间离子对数量(Number of intermolecular ion pairs)
这些属性可以根据需要进行计算和分析,以帮助研究人员更好地理解和预测分子的性质和行为。
from rdkit import Chem
from rdkit import DataStructs
from rdkit.Chem import AllChem
from rdkit.Chem import Descriptors
from rdkit.Chem import rdMolDescriptors
from rdkit.Chem import Draw
from rdkit.Chem.Draw import SimilarityMaps
## 分子量计算
mol = Chem.MolFromSmiles('c1ccccc1C(=O)O')
# 计算分子的相对分子质量
mol_weight = Descriptors.MolWt(mol)
# 输出结果
print('分子的相对分子质量为:', mol_weight)
## 分子的相对分子质量为: 122.12299999999998
## 脂溶性(LogP)
logp_m = Descriptors.MolLogP(mol)
print("脂溶性", logp_m)
## 拓扑极性表面积(Topological polar surface area)
tpsa_m = Descriptors.TPSA(mol)
print("拓扑极性表面积:", tpsa_m)
## 分子电荷分布计算
## Gasteiger电荷是一种计算分子中原子电荷分布的方法,它基于分子中每个原子周围的电子密度和邻居原子的电荷。
AllChem.ComputeGasteigerCharges(mol)
charge_atm0 = float(mol.GetAtomWithIdx(0).GetProp('_GasteigerCharge'))
print('分子第一个原子电荷', charge_atm0)
# the gasteigerCharge of the first atom - 0.04769375004654255
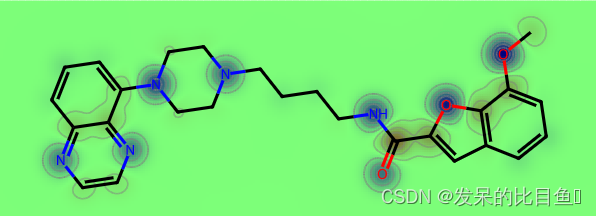
原子对性质的贡献可视化
相似性地图也可用于性质的可视化,只要性质可以分解到原子上就可以进行可视化
AllChem.ComputeGasteigerCharges 是一个函数,它可以计算分子的Gasteiger电荷。这些电荷是基于分子的共振结构和原子类型计算的,可以用于描述分子的电性、反应性和其他性质。
SimilarityMaps.GetSimilarityMapFromWeights 是一个函数,它可以根据分子中的指定特征权重生成相似性图谱。这个函数通常用于可视化分子中不同区域的相似性和差异性,以及这些区域对于分子的整体相似性的贡献。该函数需要一个RDKit分子对象和一个权重列表作为输入。
mol = Chem.MolFromSmiles("COc1cccc2cc(C(=O)NCCCCN3CCN(c4cccc5nccnc54)CC3)oc21")
AllChem.ComputeGasteigerCharges(mol)
contribs = [ float(mol.GetAtomWithIdx(i).GetProp("_GasteigerCharge")) for i in range(mol.GetNumAtoms())]
print(contribs)
d = Draw.MolDraw2DSVG(400, 400)
d.ClearDrawing()
fig = SimilarityMaps.GetSimilarityMapFromWeights(
mol,
contribs,
colorMap='jet',
contourLines=10,
draw2d=d
)
d.FinishDrawing()
with open('./data/mol29.svg', 'w+') as outf:
outf.write(d.GetDrawingText())
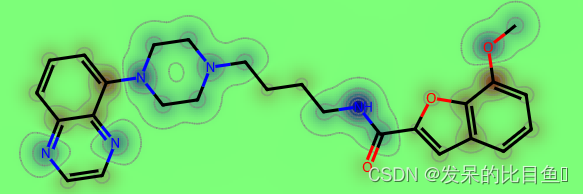
rdMolDescriptors._CalcCrippenContribs
rdMolDescriptors._CalcCrippenContribs 是一个函数,它可以计算分子的Crippen规范化贡献。这些贡献是基于分子的原子电荷和分子的几何形状计算的,可以用于描述分子的溶解度、生物利用度和其他性质。这个函数通常与RDKit分子对象一起使用。
rdMolDescriptors._CalcCrippenContribs 函数的输出结果是一个元组,其中包含两个长度为分子中原子数的列表。第一个列表包含每个原子的Crippen贡献的平均值,第二个列表包含每个原子的Crippen贡献的标准差。这些列表可以用于进一步分析分子的性质和行为。
# RDKit中内置Crippen方法计算原子logP。
mol = Chem.MolFromSmiles("COc1cccc2cc(C(=O)NCCCCN3CCN(c4cccc5nccnc54)CC3)oc21")
contribs = rdMolDescriptors._CalcCrippenContribs(mol)
print(contribs)
d = Draw.MolDraw2DSVG(400, 400)
d.ClearDrawing()
fig = SimilarityMaps.GetSimilarityMapFromWeights(
mol,
[x for x, y in contribs],
colorMap='jet',
contourLines=10,
draw2d=d
)
d.FinishDrawing()
with open('./data/mol30.svg', 'w+') as outf:
outf.write(d.GetDrawingText())
参考
https://blog.csdn.net/qq_36801966/article/details/107028297?spm=1001.2014.3001.5502



评论(0)
您还未登录,请登录后发表或查看评论